UPDATE: On March 15 2017, criteria and classifications of The Ehlers Danlos Syndromes were updated for the first time in 20 years. In light of this, I will update my guide (with the new information made available) to highlight new diagnostic criteria and classifications. You can read more about the changes here.
Because there are now 13 types of EDS, I have only covered Hypermobile Ehlers Danlos Syndrome (hEDS), Vascular Ehlers Danlos Syndrome (vEDS) and Classical Ehlers Danlos Syndrome (cEDS). If you would like me to do another guide to the rarer types, please comment below or email me. I would be more than happy to oblige!
“You’re suffering from Fibromyalgia!” “You’re depressed!” “You’re imagining it!”
“You’re malingering!” “You’re attention seeking!-”
“No I’m not – I have an Ehlers Danlos Syndrome!”
The Ehlers Danlos Syndromes (EDS) are a group of conditions that are poorly understood, even by many in the medical professions. It is essentially a defect in the production of collagen, an essential component of connective tissue.
Many articles about EDS contain medical terminology that can be difficult to understand. The purpose of this guide is to put the medical terminology in plain language and help non-affected family and friends understand exactly how EDS affects people and their day-to-day lives. The medical terminology is included in italics. Links to web pages are included throughout the article if you want to conduct your own research.
Why are they called The Ehlers Danlos Syndromes (EDS)?
The name of the condition itself is quite a mouthful! Ehlers Danlos Syndrome (Eylerz-Dan loss Sin-drome) is named after the two physicians, Dr Ehlers and Dr Danlos, who first described this group of connective tissue disorders.
What is EDS?
People with a type of EDS will produce faulty collagen. Collagen is essential for healthy connective tissue, which is found throughout the body supporting and connecting the different types of tissues and organs, including tendons, ligaments, blood vessels, internal organs, bones, the blood and skin.
Imagine a healthy person’s connective tissue as being like regular household glue. People with EDS have collagen that is more like chewing gum; stretchy and not very good at keeping things in place.
What causes EDS?
There are a number of different genes responsible for making collagen and connective tissue, so there are different types of EDS depending on which genes are faulty. There are 13 types of The Ehlers Danlos Syndromes
How did I get a faulty gene?
It is possible that the faulty gene may have been inherited from one parent, or both parents, or not inherited at all. It may be that the defect has occurred in that person for the first time. This happens in 25% of cases.
How I explained it to my 7-year old son.
A carpenter makes a wooden chair. Instead of using wood glue to place the joints of the chair together, he uses chewing gum. Once finished, the chair looks fine. But, as time goes by and the chair is used, the chewing gum doesn’t work very well at keeping the joints together. Without proper glue the chair can begin to get wobbly. I went on to explain that with proper exercise he could help to strengthen his muscles so that they acted like binding around the joints to help support them.
What does EDS feel like?
Having an EDS feels different from person to person, depending on their type, but many describe it as having a lifelong flu. Have you ever had the flu? Do you remember how painful it was having those aches and pains in the joints and muscles? Do you remember how tired and run down you felt? That’s what it’s like for people with EDS only worse and it never goes away. In addition to the daily aches and pains people with EDS also have to deal with very painful headaches, gut issues and then of course there’s the issue of dislocation. Many EDSers can’t go a day without a joint popping out. It can happen simply by stepping off a footpath or picking up a pot when cooking. A lot of people with EDS are also affected by the weather. When it is damp or when the air pressure changes their pain can increase.
How does EDS affect people?
Because collagen is everywhere in the body, there are hundreds of ways EDS can affect people. Any two people with EDS may have very different signs and symptoms, this includes people with the same type. In som,e the condition is quite mild. For others it can be disabling. Some of the rare severe types can be life-threatening.
One of the problems with diagnosing EDS is that many diseases share the same symptoms. As a result, EDS can be easily confused with other conditions and it may be difficult for doctors to recognise. But there are ways to tell if someone may be affected by EDS and need more thorough investigation. Some of the investigations available are listed later.
The most common symptoms of EDS (hEDS and cEDS) are:
- “Double jointed” – Hypermobility: joints that are more flexible than normal.
- Loose, unstable joints that dislocate easily.
- Clicking joints.
- Joint and muscle pain
In addition there may be
- Fatigue (extreme tiredness).
- Injuring easily.
- Fragile skin that bruises and tears easily. The skin may also be stretchy.
- Digestive problems
- Dizziness and an increased heart rate after standing up. (Postural Orthostatic Tachycardia Syndrome or simply POTS for short)
- Incontinence of urine in women
Digestion.
If food in the stomach doesn’t move through the body to make its way out it may just sits in the intestines and can cause a feeling of fullness, nausea, vomiting, stomach pain, to name just a few symptoms. This condition is known as Gastroparesis. (gas-tro par-eesis).
Nervous System
Another condition than often affects people with EDS is a fault with that part of the nervous system controlling the “automatic” functions of the body; things like blood pressure, breathing, heartbeat, digestion, how hot or cold you feel and the way your organs work and so on. This is called the Autonomic Nervous System. When it doesn’t operate as it should the conditions is called Dysautonomia (Dis-auto-no-me-a). Common symptoms of this are trouble with digestion, dizziness and fainting.
Dysautonomia affecting the heart.
The most common type of Dysautonomia causes dizziness and an increased heart rate after standing up. This condition is called Postural Orthostatic Tachycardia Syndrome or simply, POTS for short.
Some sufferers have fairly mild symptoms and can continue with normal work, school, social and recreational activities. For others, symptoms may be so severe that normal life things like bathing, housework, eating, sitting upright, walking or standing can be very difficult. They may feel dizzy or even faint from doing these things.
What are the symptoms for POTS?
People with POTS experience fatigue (extreme tiredness), headaches, lightheadedness (feeling dizzy), heart palpitations (when their heart beats so hard you can hear and feel it), exercise intolerance (feel ill when exercising), nausea (feeling sick), diminished concentration (hard to concentrate), tremulousness (shaking), syncope (fainting), coldness or pain in the arms, legs, fingers and toes, chest pain and shortness of breath. People with POTS can develop a reddish purple colour in the legs when standing; this is believed to be caused by blood falling down in the body because of weak veins. The colour change subsides upon returning to sitting or lying position.
Can you tell someone has EDS just by looking at them?
The short answer is no. Some may have typically blue sclera (whites of the eyes), they may have translucent skin (see through) and you may even notice how bendy they are. But some people may have some of these things and not have EDS.
Many people with the type of EDS that affects blood vessels (Vascular Ehlers Danlos Syndrome or simply, vEDS) do have some facial characteristics. Notice in the picture below that the people have big eyes, thin nose and lips.

Can EDS kill people?
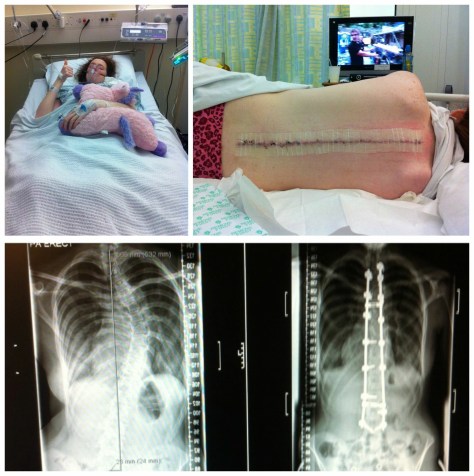
Some people think it can’t but actually, EDS has led to the untimely death of people all over the world. vEDS is considered the most serious form of EDS due to the possibility of the heart or organs tearing.
Many EDSers live a life of constant pain. This pain and misunderstanding from their medical teams, families and friends can make a person feel very sad and alone which can lead to depression and even suicide.
What treatments are available for people with EDS?
Because EDS is considered “rare” there are not many doctors willing to learn about it. Types such as hEDS and cEDS can be somewhat managed through specialised physiotherapy. Joints with weak connective tissue are more likely to dislocate. Exercises to strengthen the muscles around a joint can help stabilize the joint. Your physical therapist might also recommend specific braces to help prevent joint dislocations. Occupational therapy is also useful to help manage everyday life. Pain relief is very important for people with EDS.
EDSers should also be under the care of a Rheumatologist (a doctor who looks after bones and joints), a Cardiologist (heart doctor). There may also be a need for more specialised doctors such as Neurologists (doctors who look after the nervous system) or all of the above plus many, many more. Sometimes operations are required to repair joints that have dislocated frequently and haven’t healed properly.
Do all people with EDS need wheelchairs?
Not everyone will experience EDS the same way, some people can live normal lives and manage very well with physiotherapy and pain relief. Others may need to use wheelchairs or walking sticks to help them get around. Some people with EDS also have Gastroparesis which we discussed earlier and may need to be fed using a tube. Others may only have mild tummy problems. Some people with EDS may have to go to hospital a lot while some may only go to their GP every few months. But, just because one person can live their lives fairly normally, it doesn’t mean they don’t have EDS or that their pain shouldn’t be taken seriously.
Can you catch EDS, POTS or Gastroparesis?
No. EDS and other sub conditions are not contagious. If you know somebody with EDS, don’t be afraid, you’re not going to catch anything from them. So, if you’re avoiding someone with EDS, go make friends with them.
How can I help someone with EDS?
Be there to listen if they want to talk about it. Some people are afraid to tell you how they feel because they think friends and family don’t want to hear them complain. Ask them how they are and if you can do anything to help them. Doing shopping or household chores can be a huge help and it would be most appreciated. If you’re friend or family member has EDS and can’t access appropriate treatment like here in Ireland, write to your local representatives to tell them about EDS and the lack of care that is available. Help raise awareness in the public by sharing articles or pictures about EDS. Experts believe that EDS is not rare, just rarely diagnosed.
I will update the Diagnostic Criteria for cEDS, hEDS and vEDS in the coming days.
*Special thanks to my Dad who helped me edit this guide.*
Do you think anything else about EDS needs to be explained? Let me know in the comments!
Z.M
x